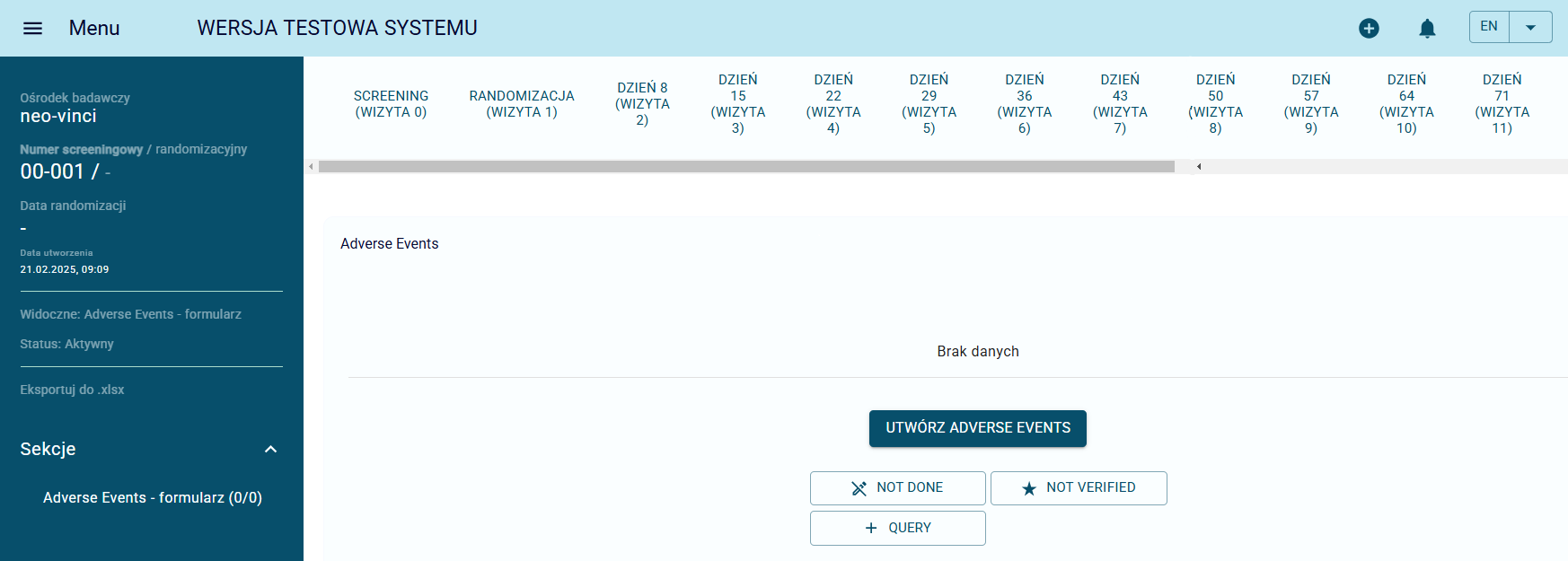
The Adverse Event (AE) Reporting Module in NEO-CRF enables fast and accurate registration and monitoring of adverse events occurring in clinical trial participants. Each AE can be reported within the system from any patient-specific form, configured according to study requirements.
A tabular view of all reported adverse events allows for efficient filtering of key information, including event type, frequency, and start date, supporting timely review and data management
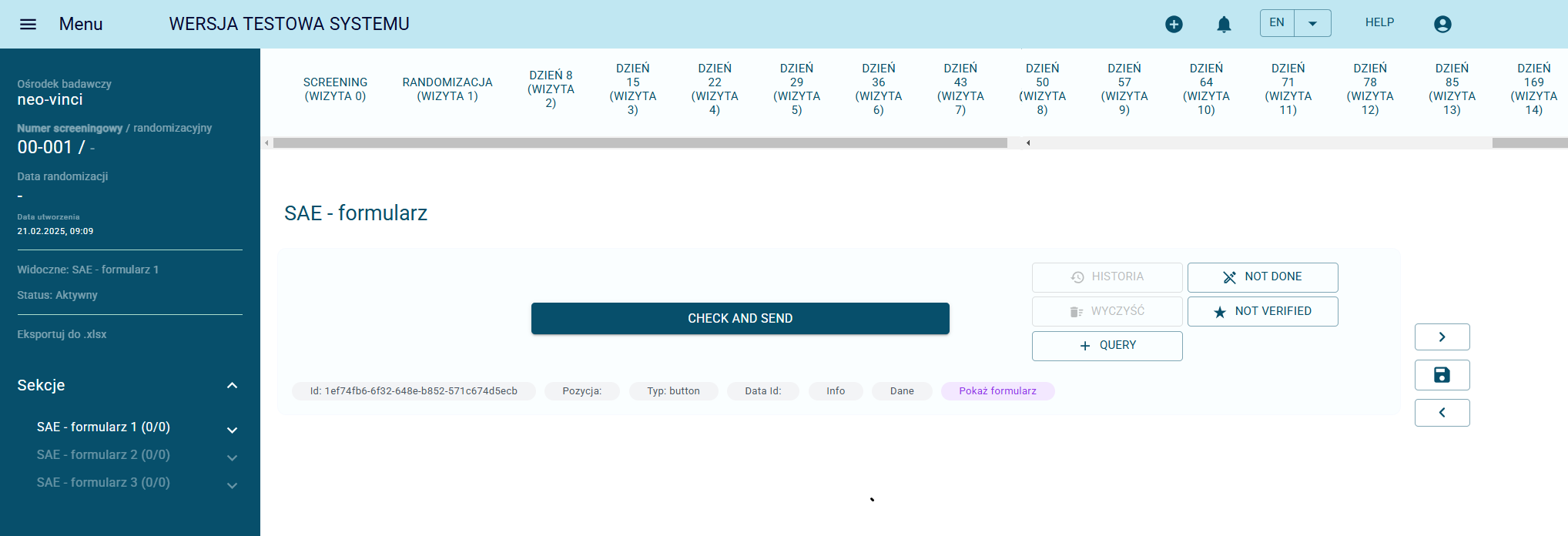
Serious Adverse Events (SAE) Reporting
In accordance with GCP guidelines, an event is classified as a Serious Adverse Event (SAE) if it results in: the patient’s death, is life-threatening, requires or prolongs hospitalization, causes permanent or significant disability, or involves an important medical event (e.g., requiring medical intervention to prevent the above outcomes).
In NEO-CRF, the SAE form is tailored to the specific needs of the study and remains fully aligned with the paper documentation. Once all required information is completed, the SAE can be submitted with a single click. For Serious Adverse Events, the system automatically send email notifications to designated users along with the exported SAE form attached.
Key advantages
Automated notifications – the system generates alerts for the Investigator, Sponsor, and monitoring team in the event of Serious Adverse Events (SAEs).
Autosave – when reporting Adverse Events, data entered into the fields is automatically saved without the need to manually click the SAVE button.
Change tracking and audit – every modification to a report is recorded, ensuring full regulatory compliance.
Simplified reporting – automatic creation of comprehensive SAE reports with all recorded event details.
Component Features
● Form structure aligned with study documentation, ● Email and SMS notifications in the event of SAE, ●User-friendly AE table layout, ● Overview of key information in one place, ● Unlimited number of reports.

 PL
PL